
On Thursday 2 May, Dr Denis Kramer, Associate Professor within Engineering and Physical Sciences at the University of Southampton, gave a seminar on novel materials for energy storage and the modelling approaches used to predict their characteristics.
This research is motivated by environmental concerns arising from our massive use of fossil fuels and their impact on the environment (amongst other things). With recent examples in Cape Town, where, due to an extremely dry summer, unprecedented freshwater shortages have hit the city in 2018. More recently in Europe, where the level of groundwater used for agriculture has reached very low levels, also due to unusually dry seasons. This motivates their research in novel energy storage materials to improve the efficiency of batteries and fuel cells. This is a vibrant area of research as investors are becoming more and more interested in investing in non-climate-damaging technologies.
The overall aim of this research is to be able to generate a comprehensive catalogue of the chemical and physical properties of different materials, where one could simply come and look for, say the best catalyst and have an answer straight after. This has been initiated with the material project (https://materialsproject.org/). This requires measuring, or more often modelling the different materials at an atomistic level, but to do this, the Schrodinger’s equation must be solved:
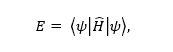
where is the energy, the Hamiltonian and psi is the wave-function, which is typically very large, even for a small system (one atom of iron has 78 electrons, this results in 1078 possible interactions, which is ridiculously large, requiring memory of approximately 1069GB (total global memory is currently 1013 – 14GB)). Solving this equation cannot be done on any realistic systems, and researchers therefore need some kind of modelling.
Density-Functional Theory
Because of the size of the wave-function, there is little hope to ever be able to solve the Schrodinger equation using standard computers. The answer may lie in quantum computing, but in the foreseeable future no large-scale quantum-computers will be available. As a result, we need to come out of this scaling problem with some clever modelling. This was done by Walter Kohn who demonstrated that it was possible to use a charged density to approximate the wave-function. This charge density can be approximated using an auxiliary quantum problem, where instead of having every electron interacting with every other electron, we have every electron interacting with the average charge density of all other electrons. This has the advantage of reducing the scaling from O(10N) to O(N3) with N the number of electrons, which is still relatively large but manageable on today’s supercomputers.

Research
Current research in Dr Kramer’s group focuses on fuel-cells and more specifically on improving the catalysts used in them. Until now, nanoparticles of platinum disposed on the surface of carbon plate were used as catalysts. But the carbon plates are prone to oxidation, and have therefore a limited lifespan and must be replaced. Investigation are currently undertaken to find new ways of creating support for this catalyst, by replacing the carbon plates with materials that will not oxide. This can easily be achieved with oxides (they will not form an oxide if they are already an oxide). In the recent years, focus has been on titanium dioxide (TiO2), but tin oxide (SnO2) can also be used. They are currently investigating if tin oxide can provide a stable support for the electric catalyst in fuel cells. Potential catalyst materials can be identified due to their position on the periodic table.
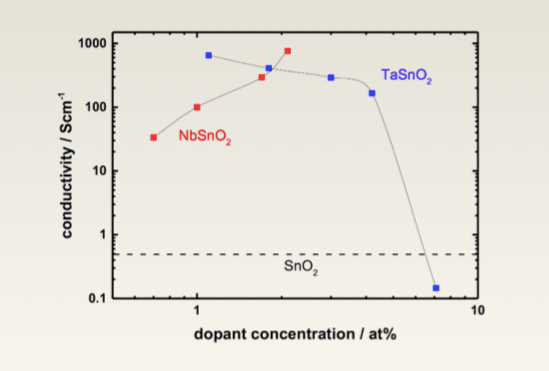
Unfortunately, oxides tend to be very good insulators, for example SnO2 has a bandgap (this is the energy required to make one electron move one “layer” of material) of 3.6eV. This redirected their research to making tin oxide more conductive. This is investigated numerically thanks to the DFT method described earlier, which allows then to derive properties of materials numerically and investigating the effect of the concentration of various dopants on the conductivity of tin oxide. Figure 1 displays tin oxide doped with Niobium (NbSnO2) and Tantalum (TaSnO2), from this figure one can see that conductivity increases with an increasing Niobium concentration and the inverse occurs as when doped with Tantalum.
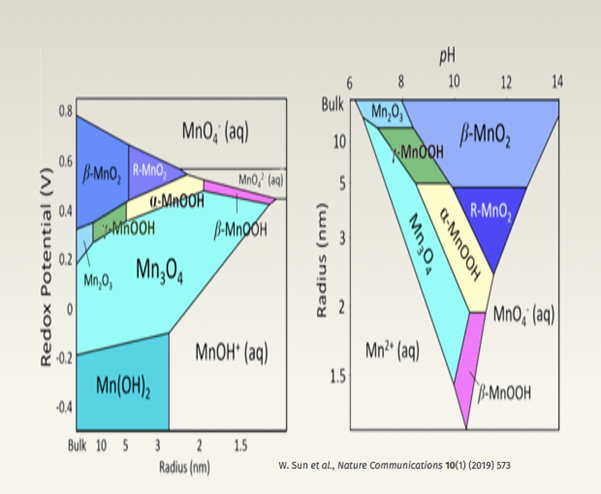
Dr Denis Kramer’s research is working towards predictive synthesis. Many factors can affect material properties; Surface energies for example are dependent on the environment in which they are synthesised. For example, various morphologies of oxide layers can be achieved by varying oxygens chemical potential through the synthesis conditions. Another example involves manganese, figure 3 (left) displays the redox potential for various crystallisations pathways during hydrothermal synthesis, whilst figure 3 (right) displays the radius of the synthesised manganese alloys as a function of pH. To briefly sum the results, the redox potential depends on the radius of the alloy, whilst the radius of the alloy depends on the pH, therefore the redox potential ultimately depends on the pH of the environment during the hydrothermal synthesis.
What’s Next
A collaboration with UCL, University of Southampton, University of Birmingham, and Loughborough is currently underway. The collaboration is funded by the EPSRC (£2.3 million), with goals of developing new energy management materials and devices. The University of Southampton is responsible for the physical vapour deposition and the high-performance computing which allows predictions, guides for synthesis and validation of the results from physical samples. Another collaboration is also on going involving the Faraday Institution, Imperial College London, University of Oxford, University of Southampton, University College London, University of Bath, University of Warwick, and Lancaster University. The aim of this collaboration is improving battery technology, as an example this improvement is achieved by reducing charge time and reducing the degradation of the cells. The research involves multi-scale time and length modelling ranging from 10-9 – 109s and 10-9 – 101m respectively. This area of research has a large industrial pull, due to this large companies such as nVidia, Ford, Siemens and CAT are involved and keen on the fruits from the research. This battery multi-scale modelling project is also funded by the EPSRC (£10 million).
Posted by Marin Lauber and Adam Murphy